Challenges for the Theoretical Description of the Mechanism and Kinetics of Reactions Catalyzed by Zeolites
J. Van der Mynsbrugge, A. T. Bell, Journal of Catalysis 2021 404 , 832-849.
DOI: 10.1016/j.jcat.2021.08.048
Abstract
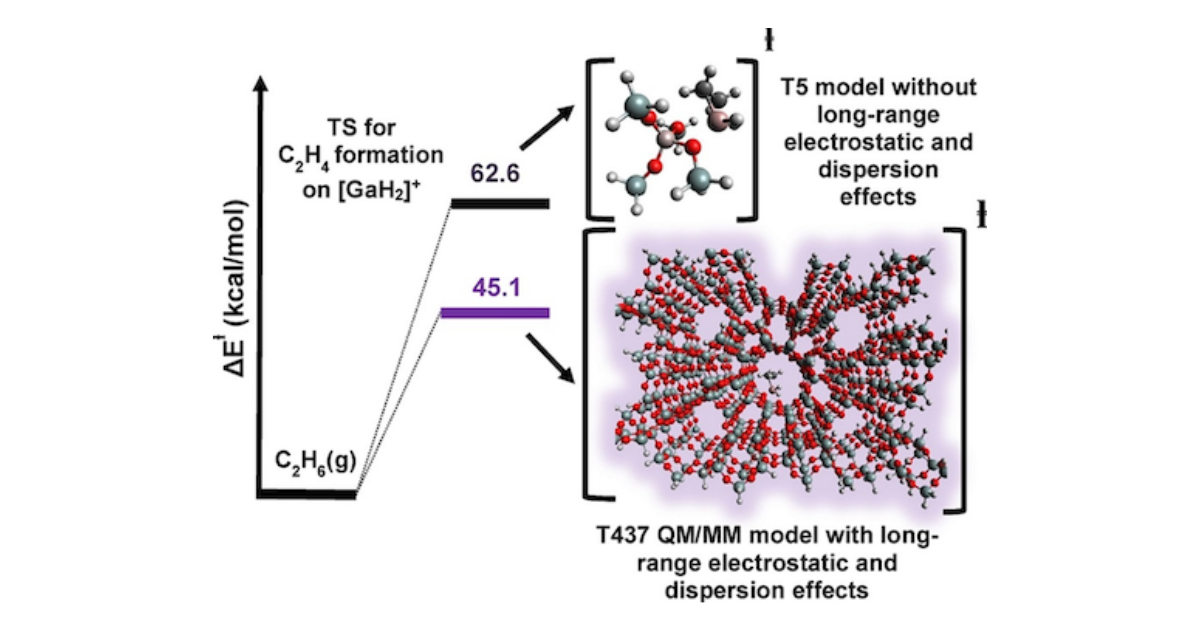
Zeolites are widely used as catalysts for the processing of petroleum to produce transportation fuels, the synthesis of a wide variety of chemicals, and for the abatement of automotive emissions. These applications have stimulated an interest in describing the mechanism and kinetics for zeolite-catalyzed reactions using theoretical methods. This Mini-review summarizes the author’s efforts towards this goal. It is shown that accurate predictions of adsorption and activation enthalpies and entropies requires that several criteria be met. The first is a correct description of the structure of the catalytically active center, as well as the portion of the zeolite framework immediately surrounding the active center and that located far from the active center. Second, the level of density functional theory (DFT) must be sufficiently high to account for the effects of dispersive interactions between the adsorbate, the active center, and the immediately surrounding zeolite atoms. Third, dispersive and coulombic interactions between the atoms in the vicinity of the active center and the balance of the zeolite framework must also be accounted for. It is shown that these conditions can be met using hybrid quantum mechanics/molecular mechanics (QM/MM) together with a high-level exchange-correlation functional and a large basis set. The success of our QM/MM approach is illustrated for reactions of light alkanes in H-MFI, as well as other protonated zeolites, and in Ga/H-MFI. We show that for low temperatures (< 400 K), the QM/MM approach gives good predictions of molecular adsorption enthalpies and activation enthalpies for elementary reactions. This is also true for higher temperatures (> 400 K) if the effects of configuration are considered using a correction obtained from configurationally biased Monte Carlo (CBMC) calculations. Calculations of the molecular adsorption entropy and the activation entropy for elementary reactions are more difficult to predict accurately. Application of the quasi-rigid rotor harmonic approximation overpredicts the loss of entropy of adsorption from the gas phase, particularly for zeolites containing large cavities and channels. CBMC corrections capture this deviation well for molecular adsorption and for early transition states resembling the adsorbed state but are inadequate for late transition states involving two loosely associated fragments.