Publications
-
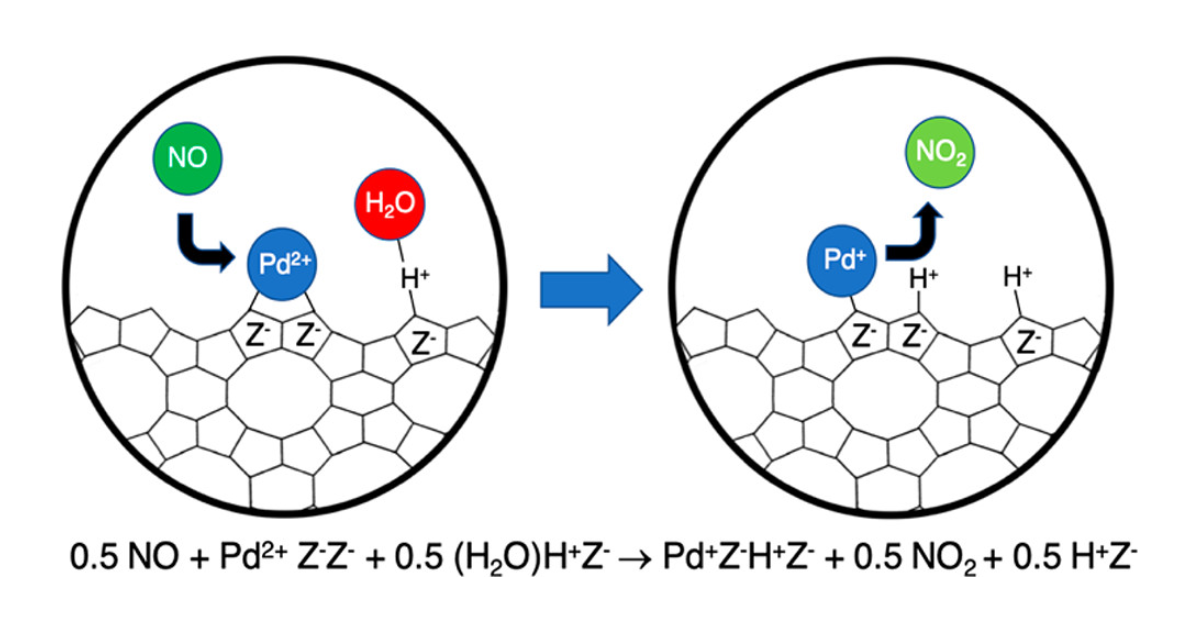
Experimental and Theoretical Studies of Pd Cation Reduction and Oxidation During NO Adsorption on and Desorption from Pd/H–CHA
P. Kim, J. Van der Mynsbrugge, M. Head-Gordon, A. T. Bell, The Journal of Physical Chemistry C 2022 126 (44), 18744-18753.
-
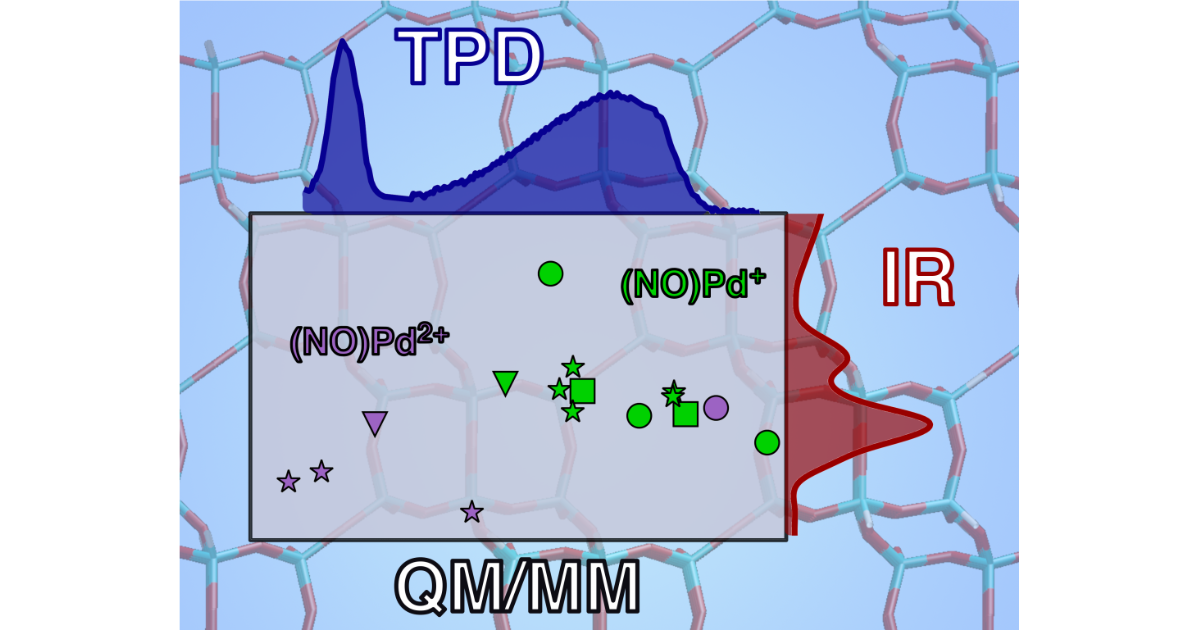
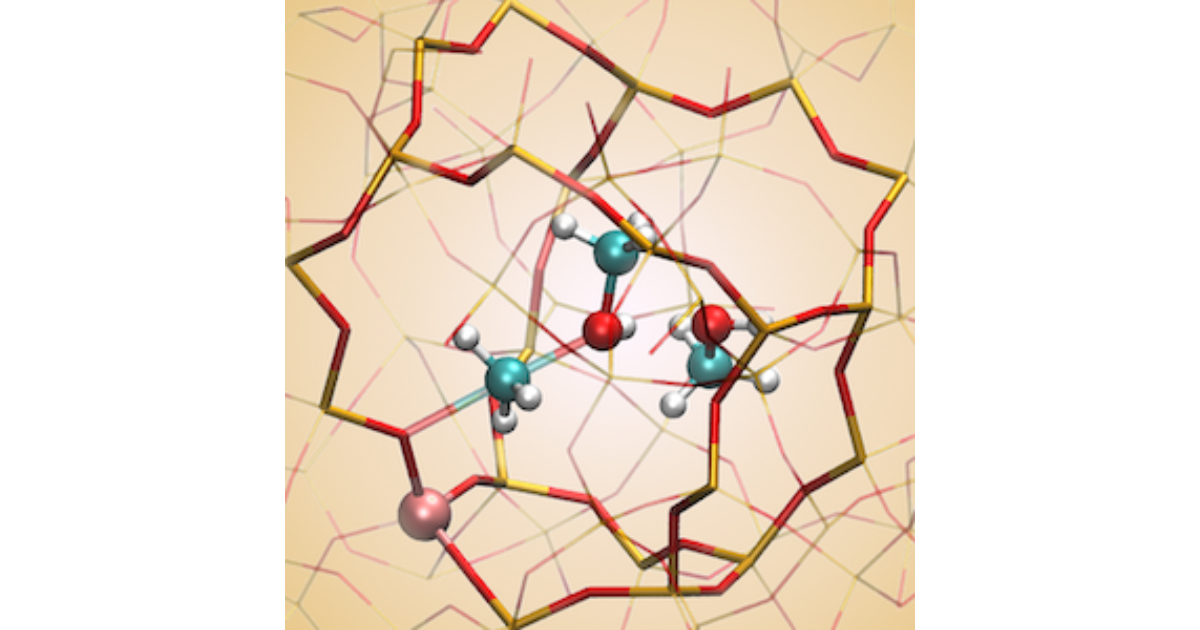
Investigation of the Modes of NO Adsorption in Pd/H-CHA
P. Kim, J. Van der Mynsbrugge, H. Aljama, T. M. Lardinois, R. Gounder, M. Head-Gordon, A. T. Bell, Applied Catalysis B: Environmental 2022 304 , 120992.
-
Challenges for the Theoretical Description of the Mechanism and Kinetics of Reactions Catalyzed by Zeolites
J. Van der Mynsbrugge, A. T. Bell, Journal of Catalysis 2021 404 , 832-849.
-
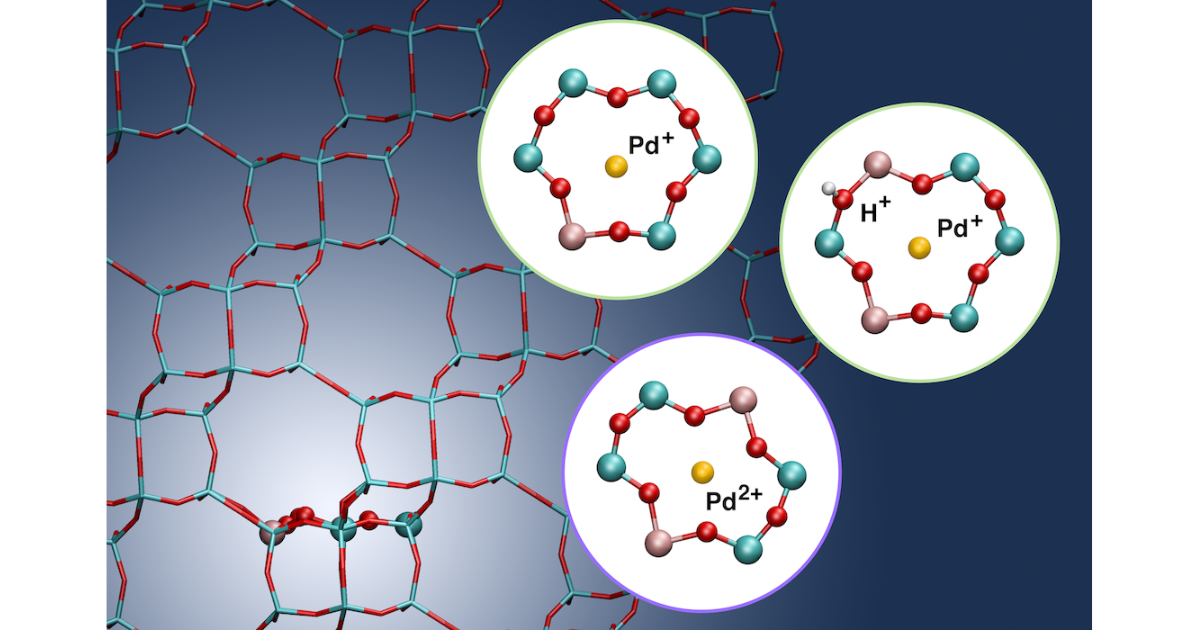
Computational Modeling Predicts the Stability of Both Pd+ and Pd2+ Ion-Exchanged into H-CHA
J. Van der Mynsbrugge, M. Head-Gordon, A. T. Bell, Journal of Materials Chemistry A 2021 9 (4), 2161-2174.
-
Facing the Challenges of Borderline Oxidation State Assignments Using State-of-the-Art Computational Methods
M. Gimferrer, J. Van der Mynsbrugge, A. T. Bell, P. Salvador, M. Head-Gordon, Inorganic Chemistry 2020 59 (20), 15410-15420.

-
Response to “Impact of Zeolite Structure on Entropic–Enthalpic Contributions to Alkane Monomolecular Cracking: An IR Operando Study”
A. Janda, L.-C. Lin, B. Vlaisavljevich, J. Van der Mynsbrugge, A. T. Bell, Chemistry – A European Journal 2019 25 (29), 7225-7226.
-
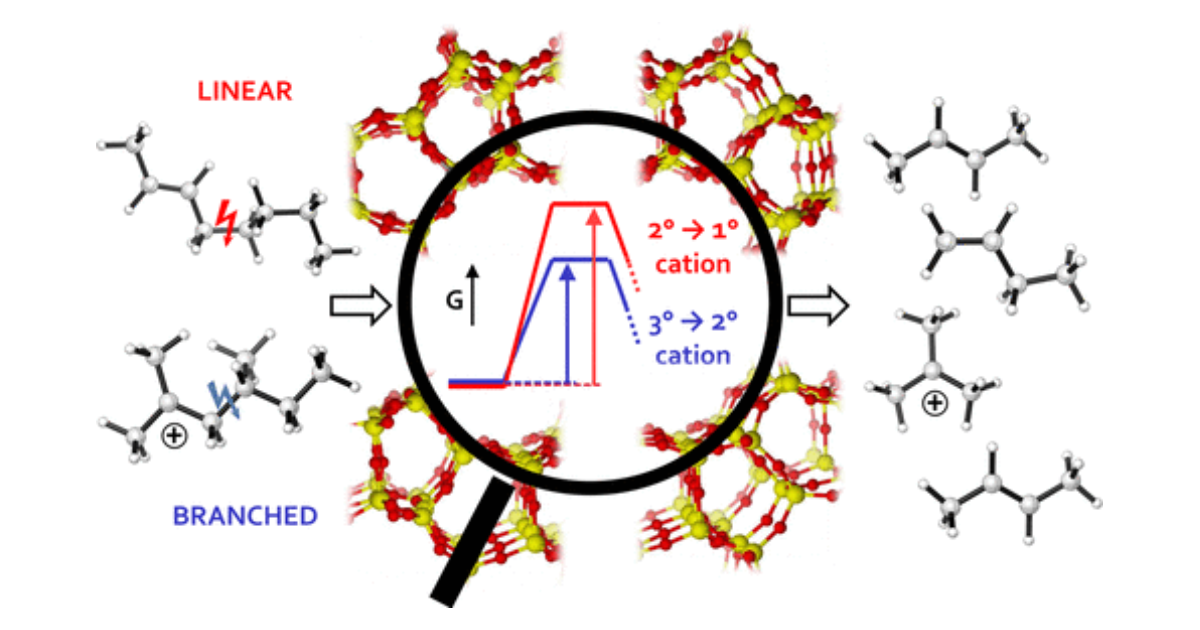
How Chain Length and Branching Influence the Alkene Cracking Reactivity on H-ZSM-5
P. Cnudde, K. De Wispelaere, L. Vanduyfhuys, R. Demuynck, J. Van der Mynsbrugge, M. Waroquier, V. Van Speybroeck, ACS Catalysis 2018 8 (10), 9579-9595.
-
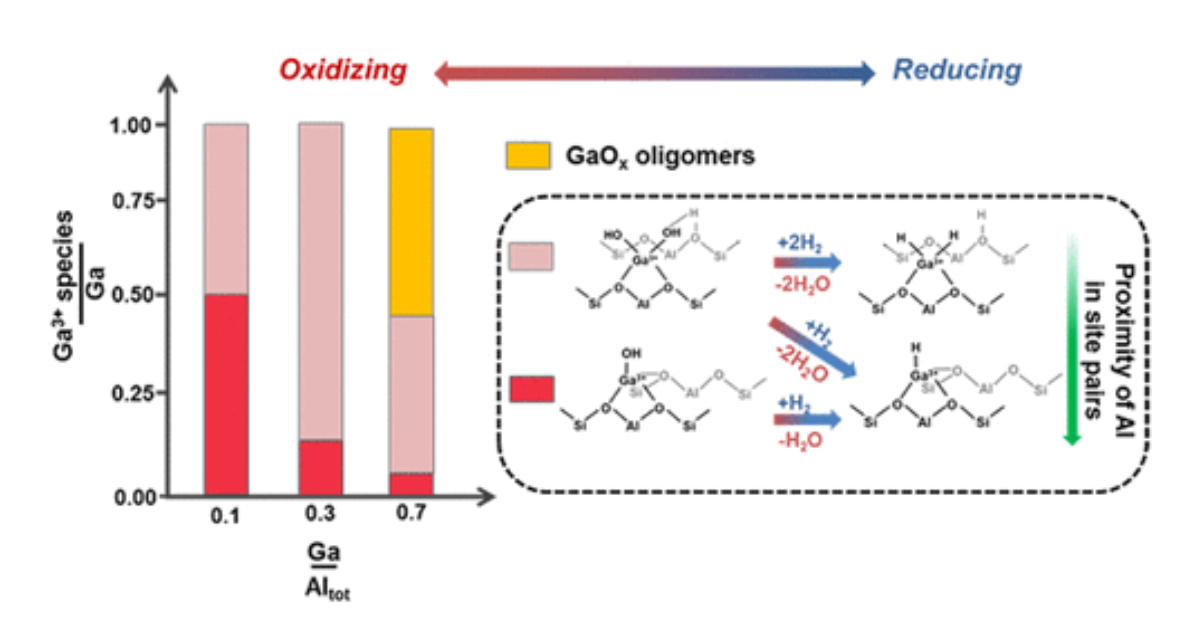
Characterization of Isolated Ga3+ Cations in Ga/H-MFI Prepared by Vapor-Phase Exchange of H-MFI Zeolite with GaCl3
N. M. Phadke, J. Van der Mynsbrugge, E. Mansoor, A. B. Getsoian, M. Head-Gordon, A. T. Bell, ACS Catalysis 2018 8 (7), 6106-6126.
-
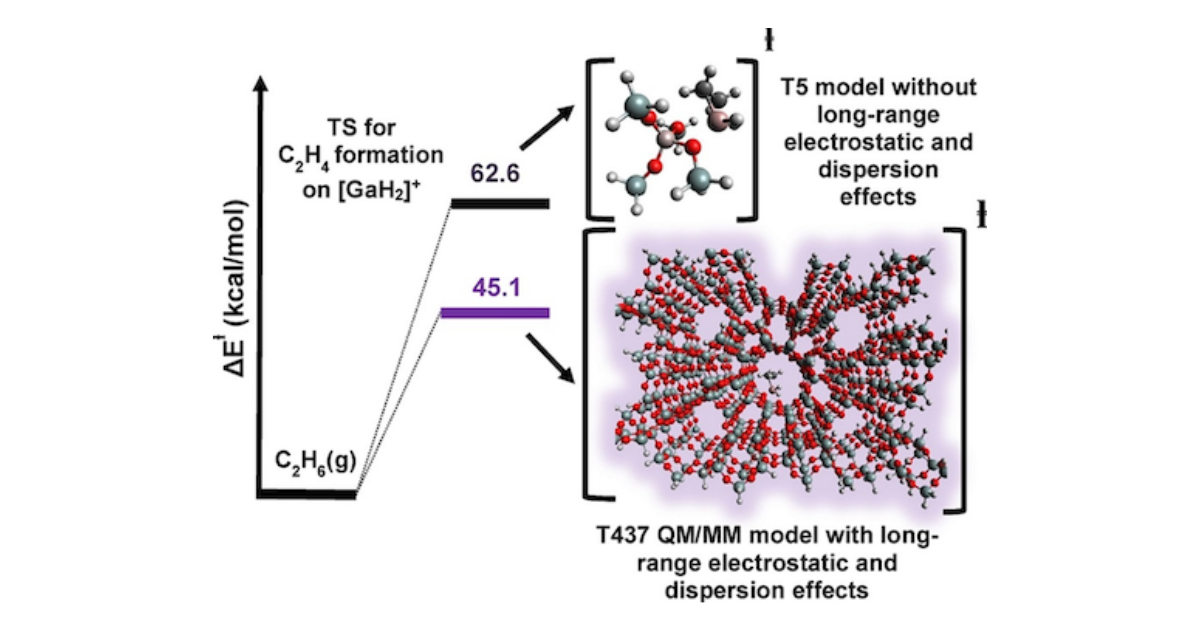
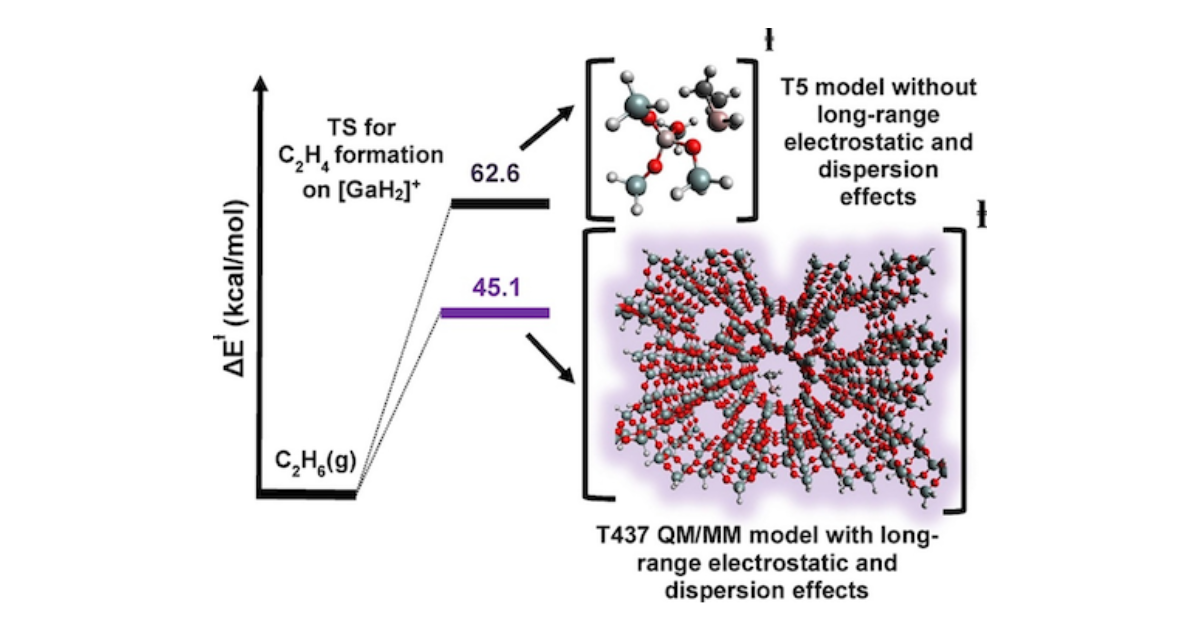
Impact of Long-Range Electrostatic and Dispersive Interactions on Theoretical Predictions of Adsorption and Catalysis in Zeolites
E. Mansoor, J. Van der Mynsbrugge, M. Head-Gordon, A. T. Bell, Catalysis Today 2018 312 , 51-65.
-
Understanding Brønsted‐Acid Catalyzed Monomolecular Reactions of Alkanes in Zeolite Pores by Combining Insights from Experiment and Theory
J. Van der Mynsbrugge, A. Janda, L.-C. Lin, V. Van Speybroeck, M. Head‐Gordon, A. T. Bell, ChemPhysChem 2018 19 (4), 341-358.

-
Theoretical Toolbox for a Better Catalytic Understanding
M. Waroquier, K. De Wispelaere, J. Hajek, S. Rogge, J. Van der Mynsbrugge, V. Van Speybroeck, in Nanotechnology in Catalysis: Applications in the Chemical Industry, Energy, Development, and Environment Protection (Eds. M. Van de Voorde, B. Sels), Wiley-VCH Verlag GmbH & Co. KGaA, 2017, 8, p. 1055-1100.

-
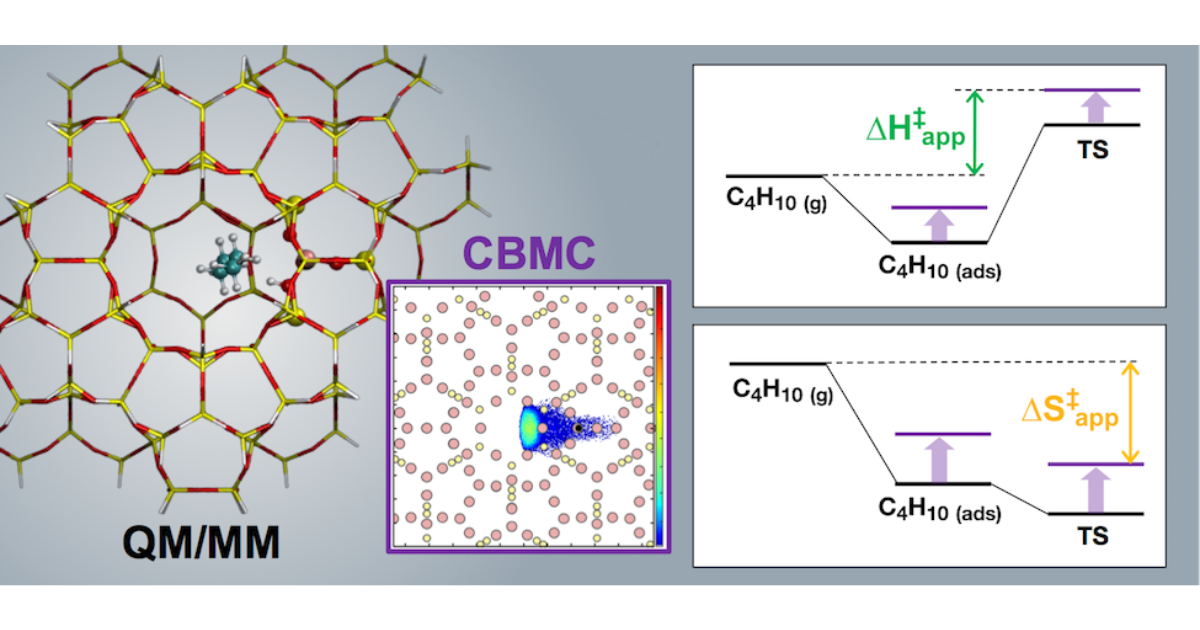
Theoretical Analysis of the Influence of Pore Geometry on Monomolecular Cracking and Dehydrogenation of n-Butane in Brønsted Acidic Zeolites
J. Van der Mynsbrugge, A. Janda, S. M. Sharada, L.-C. Lin, V. Van Speybroeck, M. Head-Gordon, A. T. Bell, ACS Catalysis 2017 7 (4), 2685-2697.
-
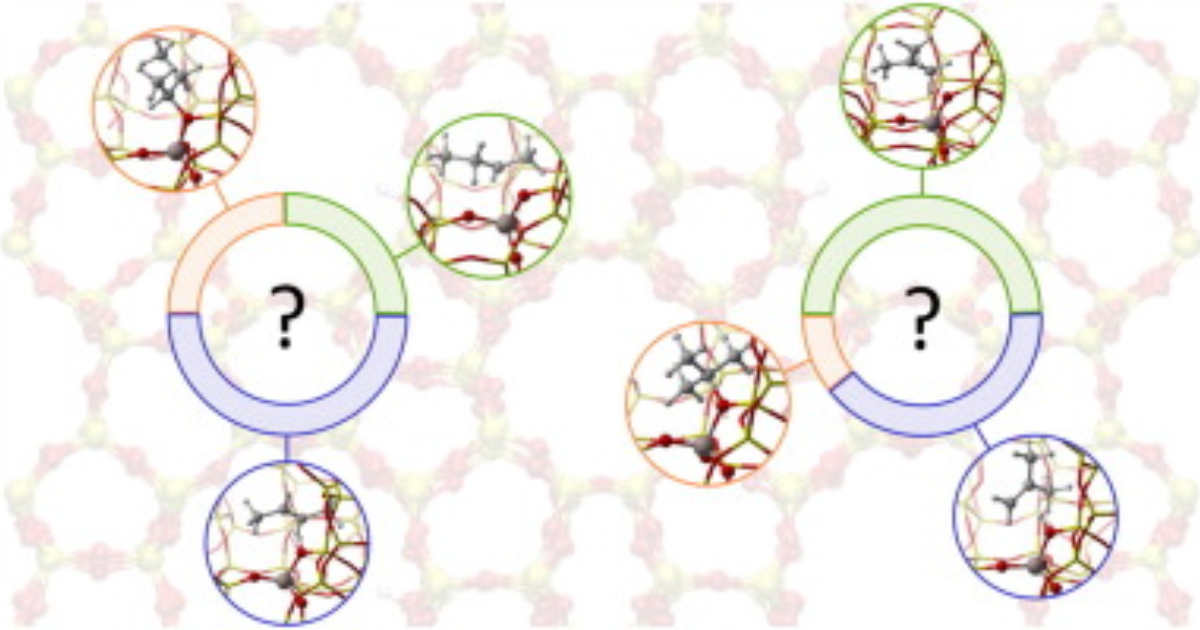
Effect of Temperature and Branching on the Nature and Stability of Alkene Cracking Intermediates in H-ZSM-5
P. Cnudde, K. De Wispelaere, J. Van der Mynsbrugge, M. Waroquier, V. Van Speybroeck, Journal of Catalysis 2017 345 , 53-69.
-
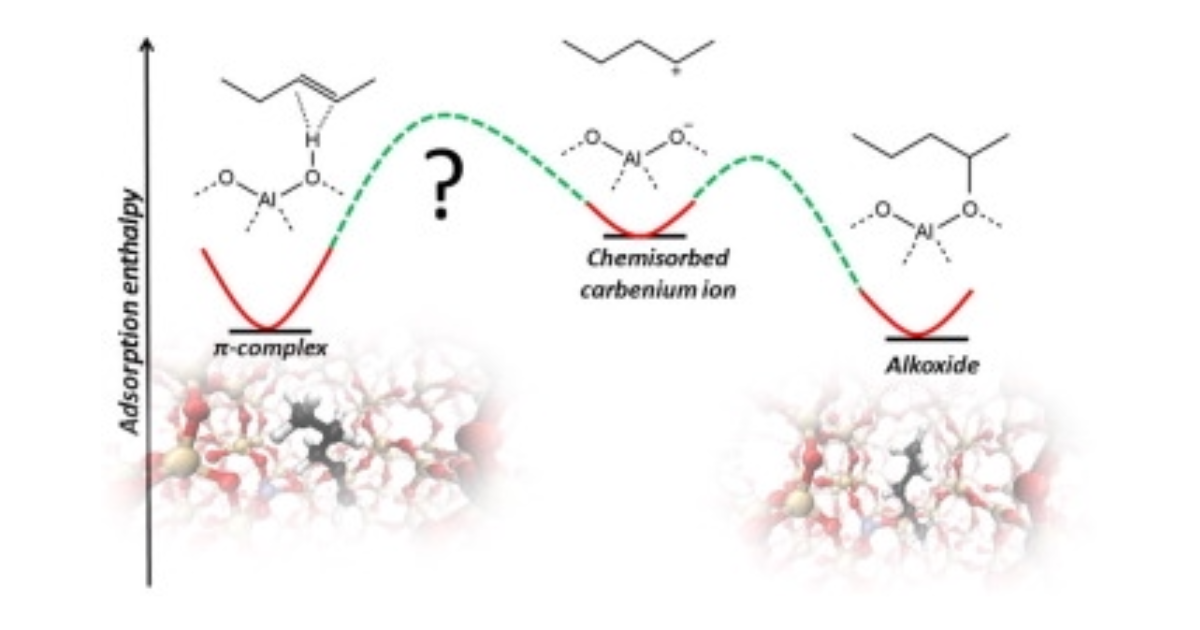
On the Stability and Nature of Adsorbed Pentene in Brønsted Acid Zeolite H-ZSM-5 at 323 K
J. Hajek, J. Van der Mynsbrugge, K. De Wispelaere, P. Cnudde, L. Vanduyfhuys, M. Waroquier, V. Van Speybroeck, Journal of Catalysis 2016 340 , 227-235.
-
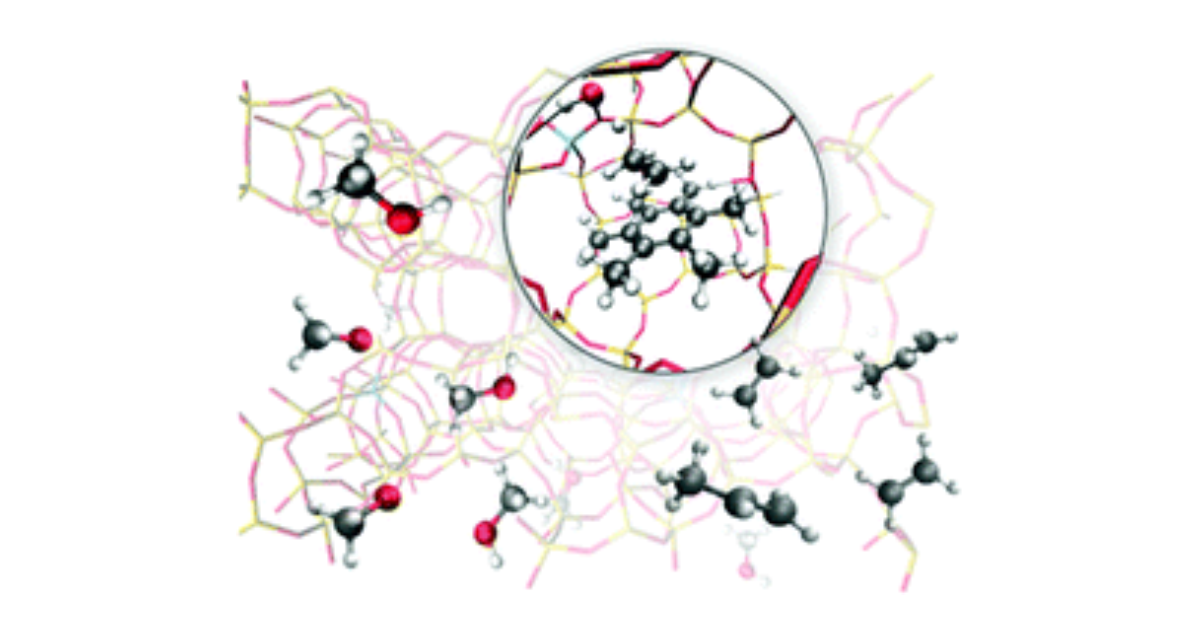
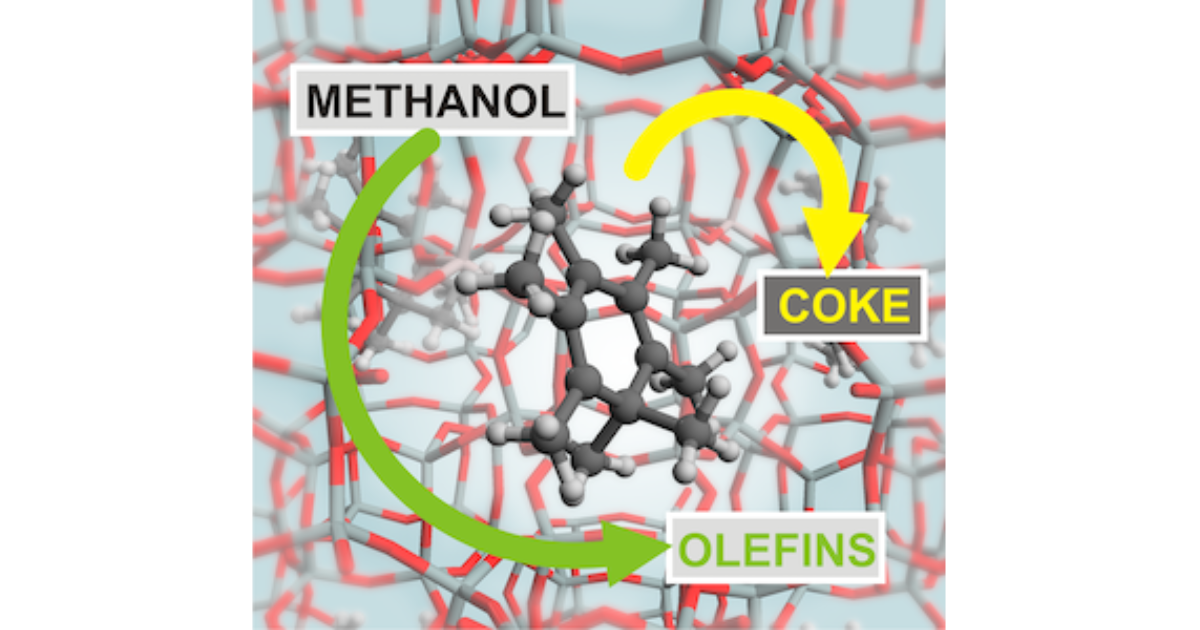
First Principle Chemical Kinetics in Zeolites: The Methanol-to-Olefin Process as a Case Study
V. Van Speybroeck, K. De Wispelaere, J. Van der Mynsbrugge, M. Vandichel, K. Hemelsoet, M. Waroquier, Chemical Society Reviews 2014 43 (21), 7326–7357.
-
Insight into the Formation and Reactivity of Framework‐Bound Methoxide Species in H‐ZSM‐5 from Static and Dynamic Molecular Simulations
J. Van der Mynsbrugge, S. L. C. Moors, K. De Wispelaere, V. Van Speybroeck, ChemCatChem 2014 6 (7), 1906-1918.
-
Molecular Dynamics Kinetic Study on the Zeolite-Catalyzed Benzene Methylation in ZSM-5
S. L. C. Moors, K. De Wispelaere, J. Van der Mynsbrugge, M. Waroquier, V. Van Speybroeck, ACS Catalysis 2013 3 (11), 2556-2567.
-
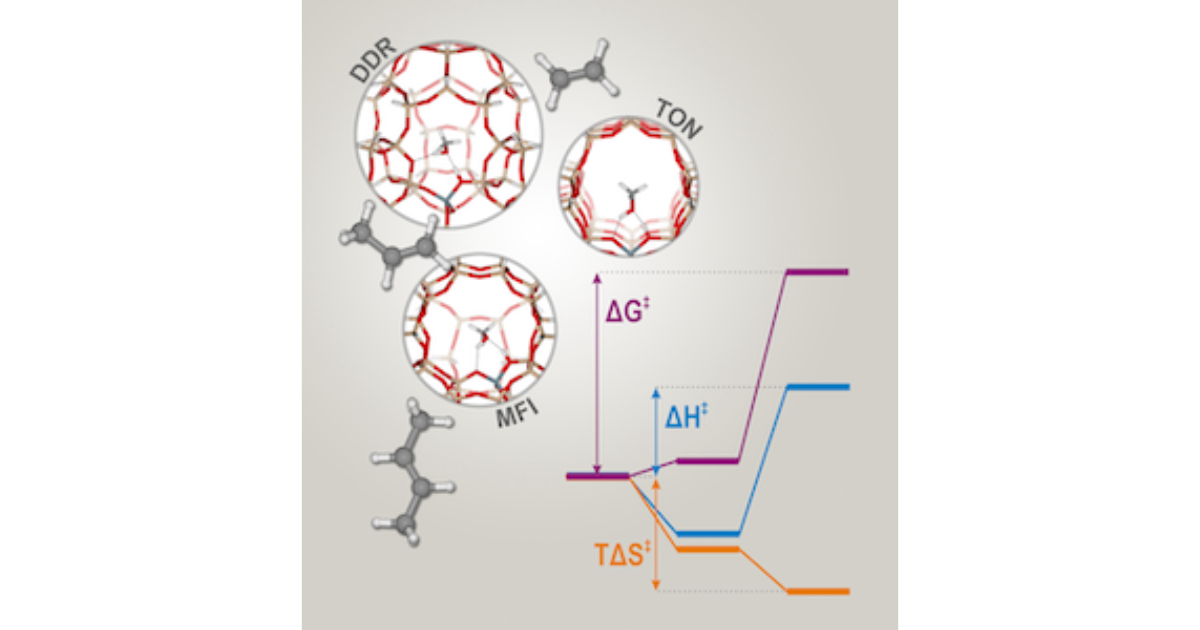
Enthalpy and Entropy Barriers Explain the Effects of Topology on the Kinetics of Zeolite‐Catalyzed Reactions
J. Van der Mynsbrugge, J. De Ridder, K. Hemelsoet, M. Waroquier, V. Van Speybroeck, Chemistry – A European Journal 2013 19 (35), 11568-11576.
-
Unraveling the Reaction Mechanisms Governing Methanol‐to‐Olefins Catalysis by Theory and Experiment
K. Hemelsoet, J. Van der Mynsbrugge, K. D. Wispelaere, M. Waroquier, V. Van Speybroeck, ChemPhysChem 2013 14 (8), 1526-1545.
-
Mechanistic Studies on Chabazite‐Type Methanol‐to‐Olefin Catalysts: Insights from Time‐Resolved UV/Vis Microspectroscopy Combined with Theoretical Simulations
V. Van Speybroeck, K. Hemelsoet, K. De Wispelaere, Q. Qian, J. Van der Mynsbrugge, B. De Sterck, B. M. Weckhuysen, M. Waroquier, ChemCatChem 2013 5 (1), 173-184.

-
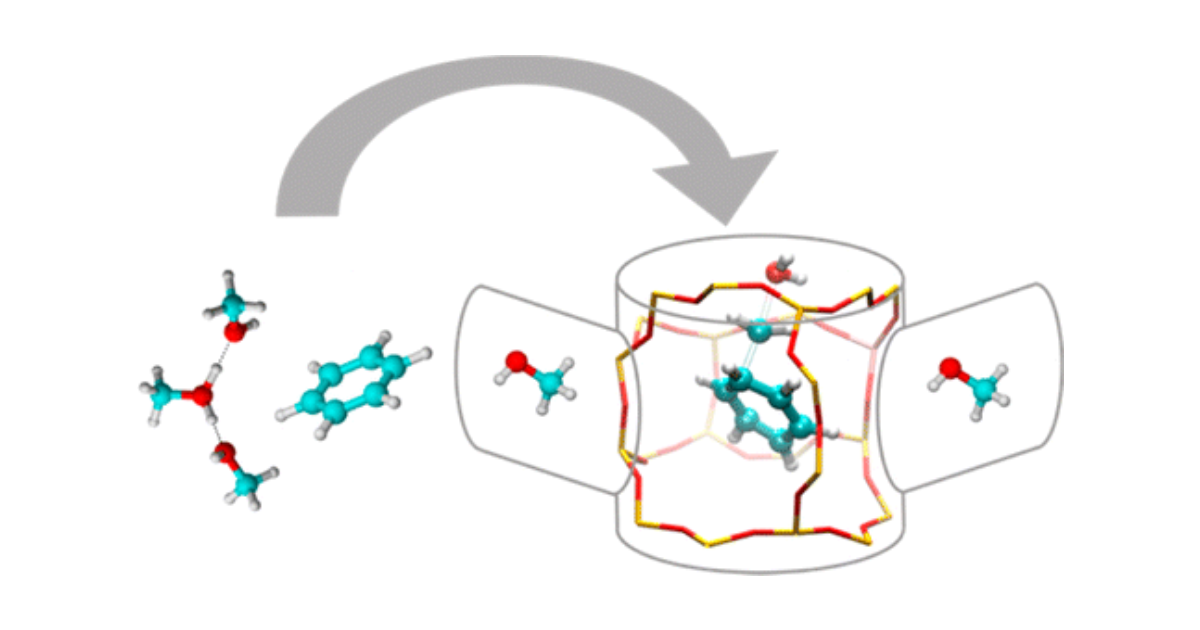
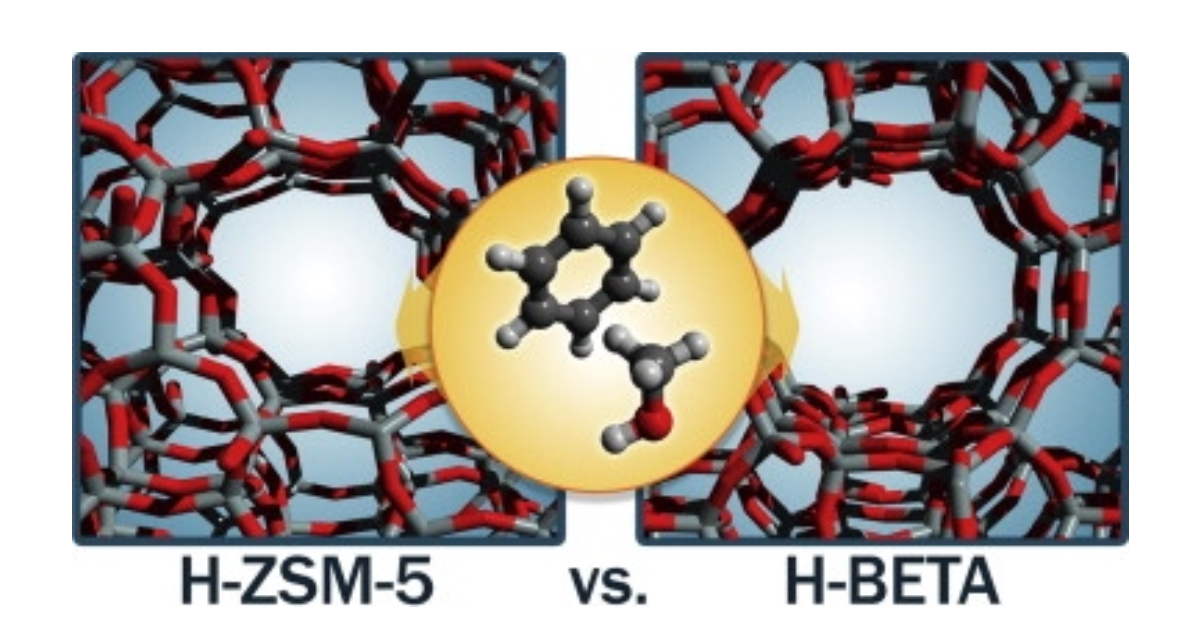
Methylation of Benzene by Methanol: Single-Site Kinetics over H-ZSM-5 and H-Beta Zeolite Catalysts
J. Van der Mynsbrugge, M. Visur, U. Olsbye, P. Beato, M. Bjørgen, V. Van Speybroeck, S. Svelle, Journal of Catalysis 2012 292 , 201-212.
-
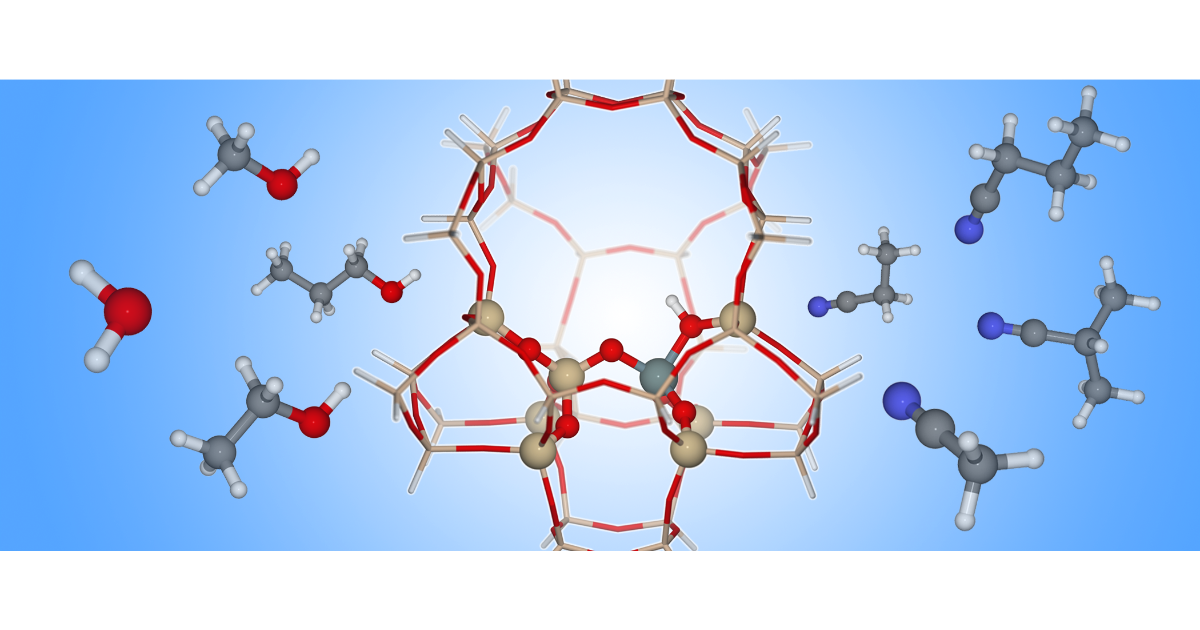
Efficient Approach for the Computational Study of Alcohol and Nitrile Adsorption in H-ZSM-5
J. Van der Mynsbrugge, K. Hemelsoet, M. Vandichel, M. Waroquier, V. V. Speybroeck, The Journal of Physical Chemistry C 2012 116 (9), 5499-5508.
-
First Principle Kinetic Studies of Zeolite-Catalyzed Methylation Reactions
V. Van Speybroeck, J. Van der Mynsbrugge, M. Vandichel, K. Hemelsoet, D. Lesthaeghe, A. Ghysels, G. B. Marin, M. Waroquier, Journal of the American Chemical Society 2011 133 (4), 888-899.
-
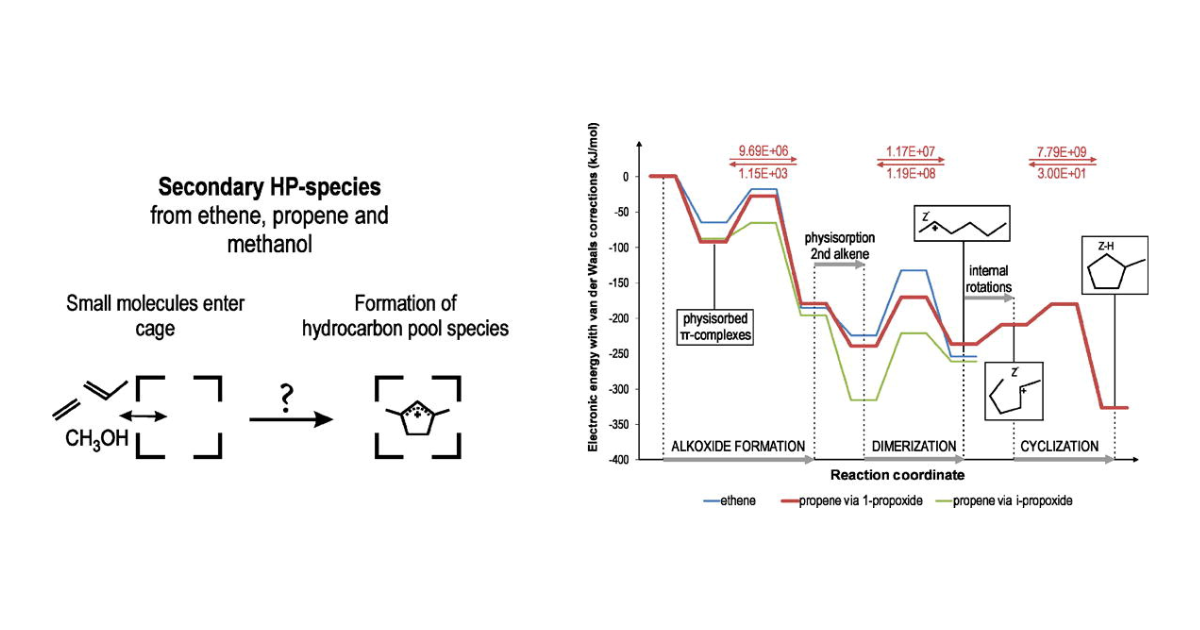
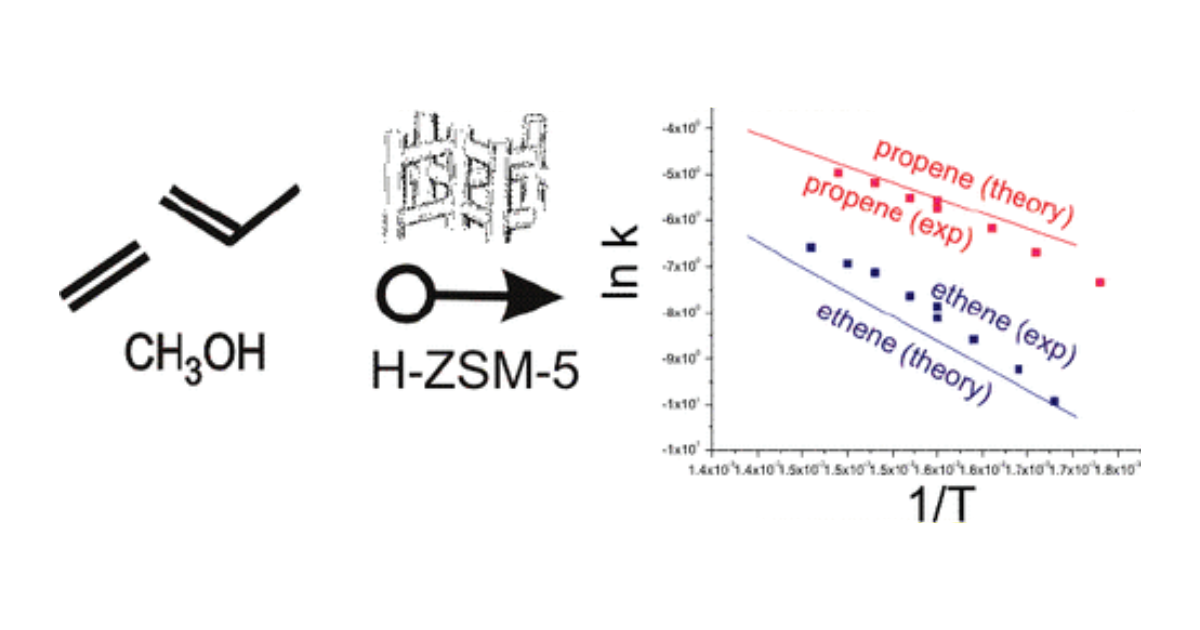
Full Theoretical Cycle for Both Ethene and Propene Formation during Methanol‐to‐Olefin Conversion in H‐ZSM‐5
D. Lesthaeghe, J. Van der Mynsbrugge, M. Vandichel, M. Waroquier, V. Van Speybroeck, ChemCatChem 2011 3 (1), 208-212.
-
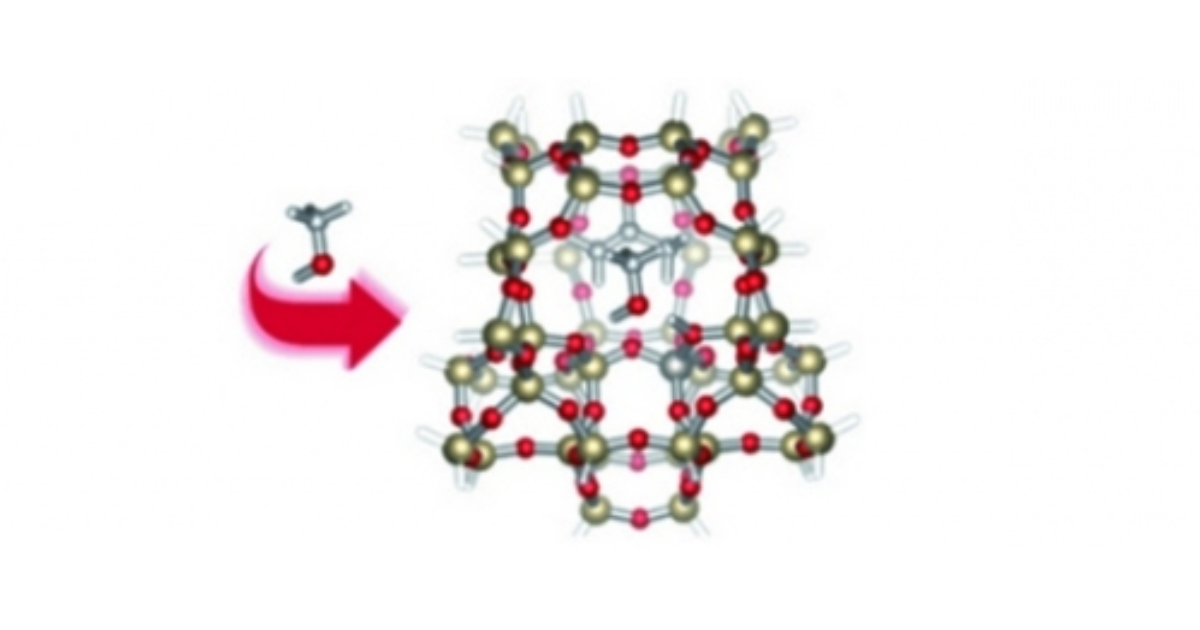
Assembly of Cyclic Hydrocarbons from Ethene and Propene in Acid Zeolite Catalysis to Produce Active Catalytic Sites for MTO Conversion
M. Vandichel, D. Lesthaeghe, J. Van der Mynsbrugge, M. Waroquier, V. Van Speybroeck, Journal of Catalysis 2010 271 (1), 67-78.